Cheminformatics Clustering
A central task in drug discovery is searching, screening, and organizing large chemical databases.
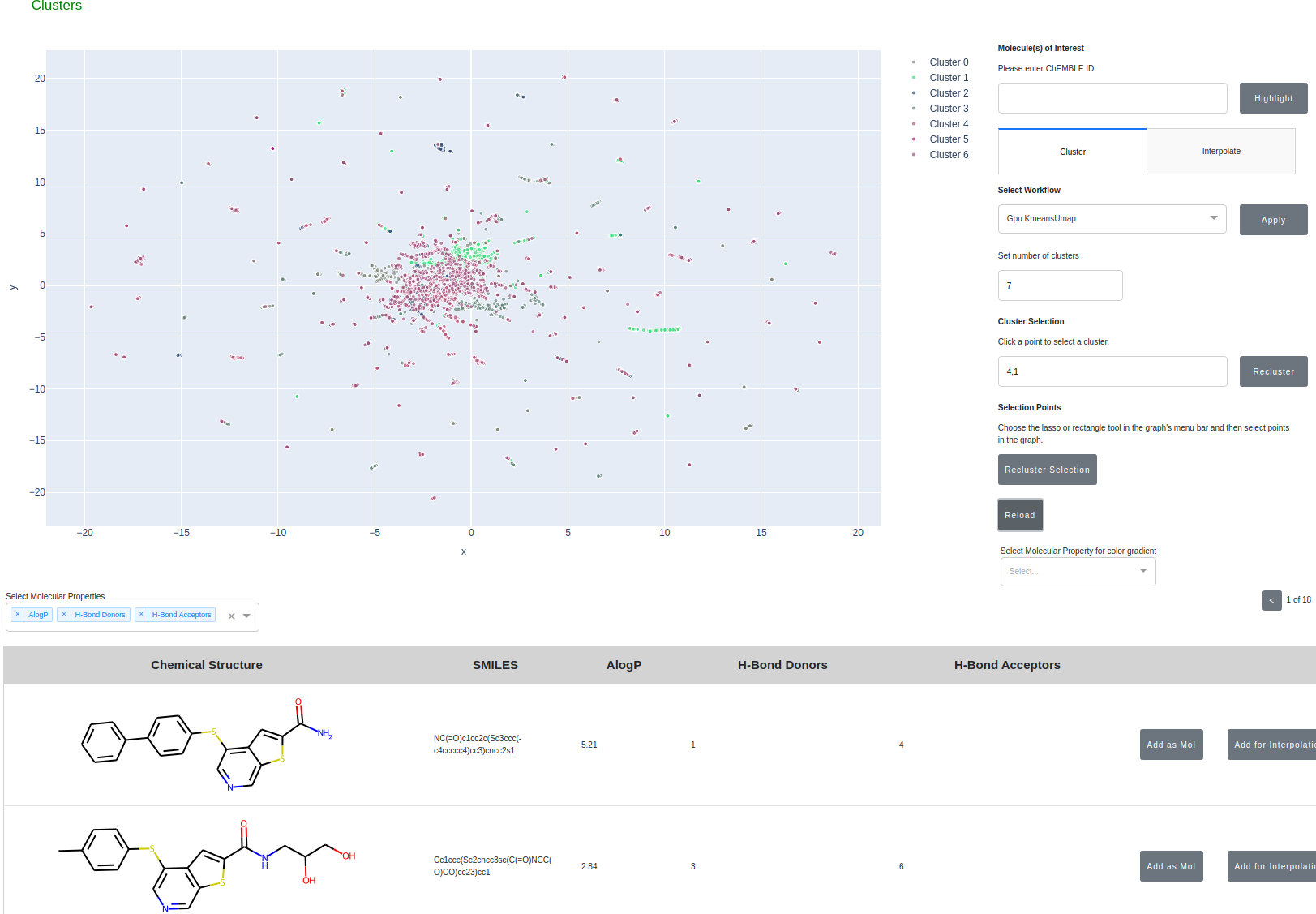
Here, we implement clustering on molecular similarity. We support multiple methods to provide a interactive exploration of chemical space.
- Compute Morgan circular fingerprints, cluster using k-means, and perform dimensionality reduction using PCA and UMAP. Distributed GPU-accelerated algorithms with multi-GPU support are used in this method. This allows processing very large datasets.
- Compute Morgan circular fingerprints, Sparse Random Projection and cluster using k-means.
- Generate new molecules either by exploring the latent space between two molecules or sampling around a molecule
Preparing the Environment (optional)
A launch script, launch.sh, is provided to perform all tasks.
Customization
The environment can be customized for control of the container, to create your own repo, or to store the data in a custom location. However, if this is not needed, skip to Getting Started to use the defaults.
To customize your local environment, edit the appropriate section of launch.sh or provide a ~/.env file with the following information below.
To generate a template for .env, just run ./launch.sh with no arguments.
If .env does not exist, then a template will be written for you.
CONT=nvcr.io/nvidia/clara/cheminformatics_demo:0.0.1
JUPYTER_PORT=8888
PLOTLY_PORT=5000
DASK_PORT=9001
PROJECT_PATH=/path/to/local/repo/dir
DATA_PATH=/path/to/scratch/space
DATA_MOUNT_PATH=/data
Getting Started
Once your environment is setup, the following commands should be all you need.
Build your container:
./launch.sh build
Download the ChEMBL database (version 27):
./launch.sh dbSetup
Launch the interactive ChEMBL exploration tool:
./launch.sh start
optional arguments:
-h, --help show this help message and exit
--cpu Use CPU
-b, --benchmark Execute for benchmark
-p PCA_COMPS, --pca_comps PCA_COMPS
Number of PCA components
-n NUM_CLUSTERS, --num_clusters NUM_CLUSTERS
Numer of clusters
-c CACHE_DIRECTORY, --cache_directory CACHE_DIRECTORY
Location to pick fingerprint from
-m N_MOL, --n_mol N_MOL
Number of molecules for analysis. Use negative numbers
for using the whole dataset.
--batch_size BATCH_SIZE
Chunksize.
-o OUTPUT_DIR, --output_dir OUTPUT_DIR
Output directory for benchmark results
--n_gpu N_GPU Number of GPUs to use
--n_cpu N_CPU Number of CPU workers to use
-d, --debug Show debug message
Navigate a browser to:
https://0.0.0.0:5000
Tutorial
See the tutorial for an example walkthrough.
Advanced Setup
Caching Fingerprints
Users can generate Morgan Fingerprints and store it in HDF5 files for later use. Please use the following command to generate fingerprint cache.
./launch.sh cache -c /data/fp
It is best to create the cache at DATA_MOUNT_PATH property defined in ~/.env. Default value of this property is /data/. This is a mounted volumne from host and available for reuse beyond the container's lifetime.
Once generated, the cached fingerprints can be used for analysis using -c option.
./launch.sh start -c /data/fp
Conda
Conda environment support is available for user wanting to use the tool outside of containers. Please find the setup file in setup directory.
Acceleration
The latest benchmarks reside in the benchmark directory.
Benchmark tests run on A100:
Benchmark tests run on V100:
Features
- Cluster molecules from ChEMBL using the embedding generated from Morgan Fingerprints --> PCA --> UMAP
- Ability to color the clustered molecules based on molecular properties
- Ability to recluster on user selected subsets of molecules or specific clusters
- Designate and track molecules of interest during the analysis
- Generate new molecules by linearly interpolating the latent space between two selected molecules or sampling arround a selected molecule
- Export generated molecules in SDF format





{kind=link}
{kind=link}